As far as I remember, it said that the data will be released on Friday on Kaggle.
I also signup to their newsletter, if I get something I can post it here.
1 Like
Thanks.
I remember something similar but didn’t remember the date. If you find the dataset on Kaggle, please let us know 
Dataset is live on Kaggle now. They’re running it as a competition
https://www.kaggle.com/c/recursion-cellular-image-classification/overview
2 Likes
A very interesting paper on how to use word embeddings to capture knowledge in the material sciences:
“Unsupervised word embeddings capture latent knowledge from materials science literature” (unfortunately behind a paywall)
What is interesting is that the used a smaller domain specific corpus instead of a huge general corpus.
This approach should transfer well to the life sciences?!
For those of you who are currently on the market, this is a great opportunity.
2 Likes
Thought I’d try to revive this thread.
Does anyone have a sense for the maximum length of the input in fastai tabular for one instance?
For example, I’d be interested in predicting the presence/absence (i.e. yes or no) of a given disease phenotype based on their DNA sample. I have labels for Yes/No for the patient phenotypes.
My thought is:
Input (DNA sample in text form) --> gene2vec autoencoder --> fast.ai NLP for binary prediction of Yes/No for phenotype.
Seems to me the bottleneck might be the volume of DNA text information per patient.
Has anyone attempted something like this using ‘raw’ genetic information?
If so, were you also able to perform feature extraction to see which components of the DNA sequence were important in arriving at the phenotype prediction?
I’ve done ULMFiT on genomic sequences (link) which has worked well for classification of sequences up to a few kb. I was also able to infer important sequence regions by perturbing regions of the sequence to see the impact on the classification confidence. Here’s an example for promoter classification, where the red line shows the true transcription start site.
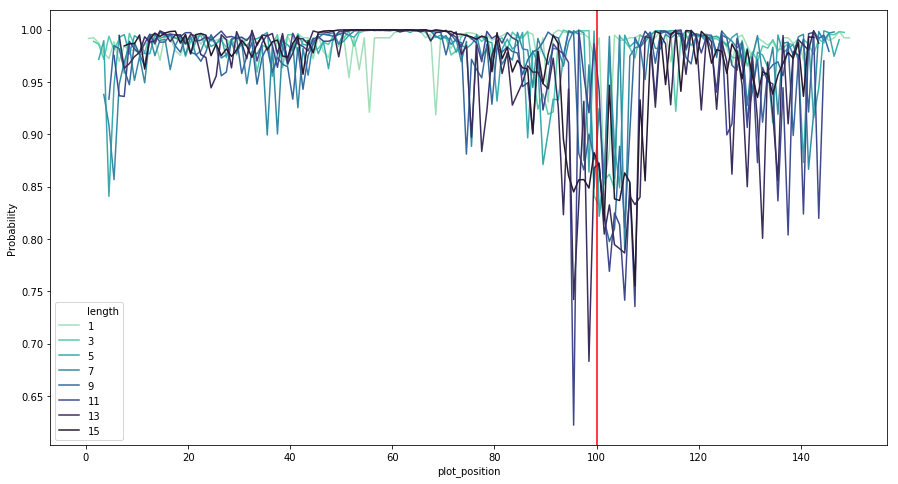
How much sequence information are you dealing with? I’ve been curious about the ability of LSTM models for genomics classification to scale to extremely long sequences, given the difficulty of maintaining information over a large number of time steps.
You might also consider looking into how SVD can be used on genomics data to reduce dimensionality and extract features in an unsupervised fashion.
2 Likes
Hi Karl,
Thanks for pointing me to your Git repository. Impressive stuff!
I am essentially looking to use an entire patients’ human genome as input. For example, we have patient whole DNA samples (extracted through tissue) of patients ‘with’ and ‘without’ the disease. In that sense, this would be a supervised classification problem as we know the outcome. But, I suspect adding in a dimensionality step prior to the supervised component will be prudent given the size of the human genome.
I figured I might treat this as an NLP problem, as perhaps there are specific and meaningful sequence ‘embeddings’ discovered that would point to a common genetic link in the diseased patients.
That’s a difficult problem due to the scale of the input. A whole genome contains a huge amount of information, most of it noise to your particular task. It’s so high dimensional I’d expect a neural network to learn a large number of features specific to the dataset that don’t generalize.
Come to think of it, I actually don’t know of any research doing whole genome analysis with deep learning. It looks like there’s a paywalled paper from a few months ago:
https://www.nature.com/articles/s41588-019-0420-0
I wonder if one way to narrow the search space would be to break the patient genomes into chunks of ~1kb, then train a classification model on the chunks. The classification model would of course perform poorly, as most 1kb chunks are not definitively indicative of disease/no disease, but you may be able to learn something by looking at what genome chunks work well for classification and what don’t. Ideally this sort of test would allow you to identify smaller regions of the genome that allow your model to make good predictions of disease/no disease.
If you could narrow down the entire genome to specific regions, you could then train a model on just those regions for predictive purposes.
1 Like
“Deep learning enables rapid identification of potent DDR1 kinase inhibitors”
https://www.nature.com/articles/s41587-019-0224-x (Unfortunately behind a paywall…)
GitHub repo:
The publication is from http://insilico.com which already published some very interesting stuff in that field.
1 Like
article
s41587-019-0224-x.pdf (2.7 MB)
suplement
41587_2019_224_MOESM1_ESM.pdf (1.9 MB)
@MicPie article that was behind paywall
1 Like
Hi Karl,
Thanks for this great resource! You said that it works well up to few kb… How about human genes - about 10-12kb on average?
Could it be used to get fixed-length embedding vectors for gene DNA sequences?
Thanks,
Sal
Thanks Karl, for developing the ULFMiT model to genomic and starting this thread ! I’m using your notebooks as a tutorial and applying to my own datasets as a way to learn more about DL and text classification. I’m also testing in some long sequences (> 20 kbp) and struggling with the results. When you pinpoint that you had to change the max_len parameter default to achieve better results, you mean the max_len parameter in function get_model_clas() or
in another function ?
Hey Rodrigo,
That would be the max_len parameter in the get_model_clas function. For some context, the max_len parameter is fed to the MultiBatchEncoder module which is part of the standard fastai library.
The MultiBatchEncoder saves the last max_len hidden states to send to the classification head. So if your max_len value is 100 but your sequence length is 1000, your classification head only sees those final 100 hidden states. This can be an issue if major features are in the start/center of your sequence.
How do we improvise a drug to make it better than the existing version using Deep Learning. How can we redesign a drug that can interfere interaction between two proteins? How we can use Deep Learning to design peptides that can interfere protein/protein interactions. Can you please point me to some good deep learning papers in this space.
Hi Bay Area people on this thread,
You might be interested in these events:
-
AI in drug discovery & medicine MIT presentations at JPM Wed Jan 15th, 6-9pm
@ $150 for 3 hours entry to this event is a little steep.
If anyone can get me a free ticket, I’d love to go! -
BayBIFX @ Thermo Fisher, FREE Tue Dec 3rd, 4-7pm.
Often presenters, organizers, and anyone else who wants to join go out for dinner afterward.
1 Like
Summary of the “Machine Learning for Health” workshop from NeurIPS 2019:
1 Like
Hi all - happy to have found this topic!
I’ve been working on a language model of DNA sequences. I’m an environmental microbiologist studying how microbes regulate the carbon cycle and the Earth system in general, and trying to build better models of the interactions between microbes and their environment.
For now, I’ve been working with raw, unassembled NGS reads from metagenomes (100-200bp long) as input to the model. I’m building a character-level model with an AWD-LSTM architecture - so tokenizing the sequence one nucleotide at a time (with a stride of one). I’ve been getting promising preliminary results when bidirectional is set to ‘False’ (accuracy in the ballpark of 0.35-0.4 in early epochs), but when I set bidirectional to ‘True’ I get 0.98 accuracy on the first epoch… Clearly it’s somehow able to cheat, but I can’t figure out how this would be. If I was using a kmer/ngram size of 3 and a stride of 1, you could see how a bidirectional model would be able to ‘see’ the right answer from the surrounding tokens and cheat that way, but for a character-level model this shouldn’t be the case… any ideas what I’m missing??
It occurred to me that the fact that the sequences are paired-end could have something to do with it - there’s overlap of maybe 30bp or so between training examples - but haven’t tested it yet. And I’m not sure why that would affect only a bidirectional model - in that case you’d think a forward-only model would also be affected.
Here’s how I’m setting up the model:
config = awd_lstm_lm_config.copy()
config['bidir'] = True #or False
#'data' is my databunch
learn = language_model_learner(data, AWD_LSTM, drop_mult=drop_mult, model_dir=".", config=config, pretrained=False)
learn.to_fp16()
learn.callbacks.append(SaveModelCallback(learn, every='improvement', monitor='accuracy', name=model_path))
learn.callbacks.append(CSVLogger(learn, filename=log_path, append=True))
learn.fit_one_cycle(numcycles,lrate, moms=(0.8,0.7))
Thanks all!
3 Likes
Have a look here for a great starting intro:
I’m not sure if these repos also looked into using SentencePiece, but this should be now easier with fastai and could be very promising.
Keep us updated on how your approach turns out!
3 Likes
You can’t use bidirectional LSTMs for language models out of the box. Language models predict the next token for every token in the input. If your model is bidirectional, it reads the target token as part of the input.
If you want to use bidirectional LSTMs for language modeling, you’ll have to train your model with masking like BERT and other transformer based language models do.
1 Like